

Cumulus is a cloud-based single-cell genomics and spatial transcriptomics data analysis framework that is scalable to massive amounts of data, cost-effective, and able to process a variety of data types. It is cloud natively developed, leverages the Pegasus library and many other open-source software (e.g. cellranger/starsolo), and is publicly available in GitHub as well as pre-installed in Terra (the BCDC cloud computing environment). It supports analysis of single-cell RNA-seq, CITE-seq, Perturb-seq, single-cell ATAC-seq, single-cell immune repertoire and spatial transcriptomics data.
RRID: SCR_021644
Generic Web URL: https://cumulus.readthedocs.io/en/stable
Terra URL: https://app.terra.bio/#workspaces/kco-tech/Cumulus
Contact: cumulus-support@googlegroups.com
Loom is an efficient file format for very large omics datasets, consisting of a main matrix, optional additional layers, a variable number of row and column annotations, and sparse graph objects. Loom files are portable, self-contained and ensure that metadata travels with the data. Under the hood, Loom files are HDF5 and can be opened from many programming languages, including Python, R, C, C++, Java, MATLAB, Mathematica, and Julia. The Loom file format is natively supported by popular scRNA-seq packages including Scanpy, Seurat, SCope and scVI. (LoomPy, RRID:SCR_016666)
Access Loom here:
https://loompy.org
S. Linnarson Lab (http://linnarssonlab.org/), Karolinska Institute
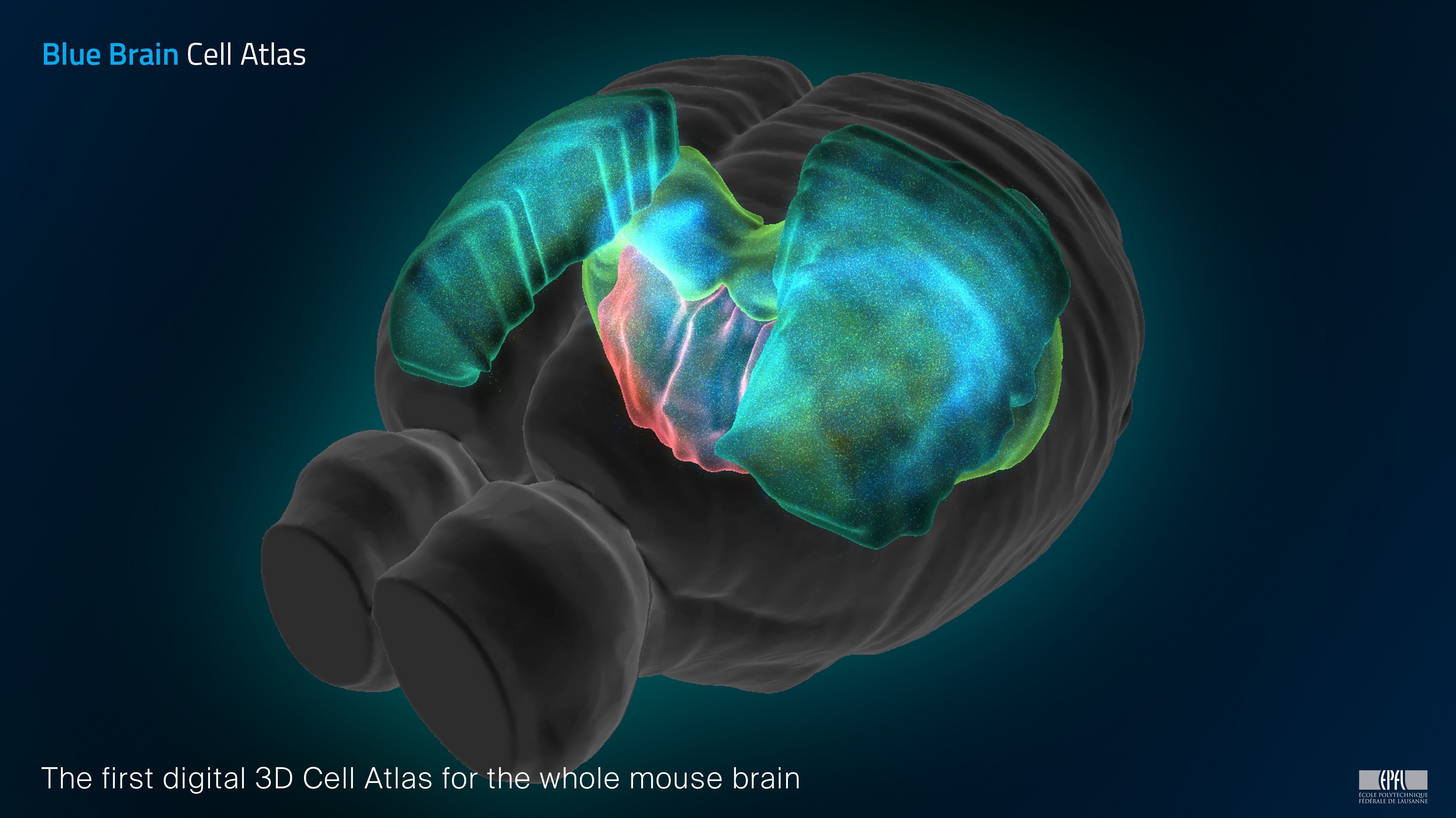
The Blue Brain Cell Atlas is a comprehensive online resource that describes the number, types, and positions of cells in all areas of the mouse brain. The atlas provides the densities and positions of all excitatory, inhibitory and neuromodulatory neurons, as well as astrocytes, oligodendrocytes and microglia in each of the brain regions defined in the Allen Mouse Brain Atlas. Users can download cell numbers for statistical analysis, cell positions and types for modeling and visualizing brain areas. The underlying workflow uses imaging data from the Allen Institute Common Coordinate Framework to generate cell positions and assign their type using the API for data access. (Blue Brain Project, RRID:SCR_002994)
Access the Blue Brain Cell Atlas here:
https://bbp.epfl.ch/nexus/cell-atlas/
Blue Brain Project (https://bluebrain.epfl.ch/), EPFL
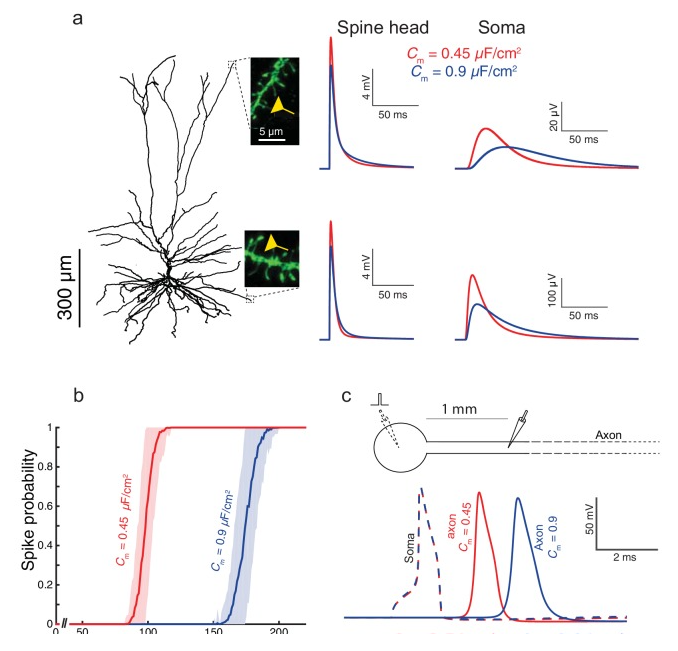
ModelDB, Biophysical Models - The advanced cognitive capabilities of the human brain are often attributed to our recently evolved neocortex. However, it is not known whether the basic building blocks of human neocortex, the pyramidal neurons, possess unique biophysical properties that might impact on cortical computations. The Segev group has shown that layer 2/3 pyramidal neurons from human temporal cortex (HL2/3 PCs) have a specific membrane capacitance (Cm) of ~0.5 µF/cm2, half of the commonly accepted “universal” value (~1 µF/cm2) for biological membranes. This finding was predicted by fitting in vitro voltage transients to theoretical transients then validated by direct measurement of Cm in nucleated patch experiments. This is the first demonstration that human cortical neurons have distinctive membrane properties, suggesting important implications for signal processing in human neocortex. They also have developed detailed models of pyramidal cells from human neocortex, including models on their excitatory synapses, dendritic spines, dendritic NMDA- and somatic/axonal Na+ spikes that provided new insights into signal processing and computational capabilities of these principal cells. (ModelDB, RRID:SCR_007271)
Access the related ModelDB models and codes here:
https://senselab.med.yale.edu/ModelDB/showmodel.cshtml?model=195667#tabs-1
https://senselab.med.yale.edu/ModelDB/showmodel.cshtml?model=238347#tabs-1
I. Segev Lab (https://elsc.huji.ac.il/faculty-staff/idan-segev), Hebrew University
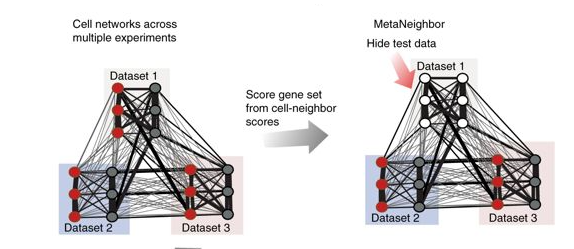
MetaNeighbor quantifies the degree to which cell types replicate across datasets, and enables rapid identification of clusters with high similarity. MetaNeighbor first measures the replicability of neuronal identity, comparing results across eight technically and biologically diverse datasets to define best practices for more complex assessments. By taking the correlations between all pairs of cells a network is built where every node is a cell and the edges represent how similar each cell is to each other cell. This network can be extended to include data from multiple experiments (multiple datasets). To assess cell-type identity across experiments neighbor voting is used for cross-validation, systematically hiding the labels from one dataset at a time for testing. Cells within the test set are predicted as similar to the cell types from other training sets using a neighbor-voting formalism. Whether these scores prioritize cells as the correct type within the dataset determines the performance, expressed as the AUROC. Comparative assessment of cells occurs only within a dataset, but is based only on training information from outside that dataset. (MetaNeighbor, RRID:SCR_016727)
Access MetaNeighbor here:
https://www.bioconductor.org/packages/release/bioc/html/MetaNeighbor.html
J. Gillis Lab (http://gillislab.labsites.cshl.edu/), Cold Spring Harbor Laboratory
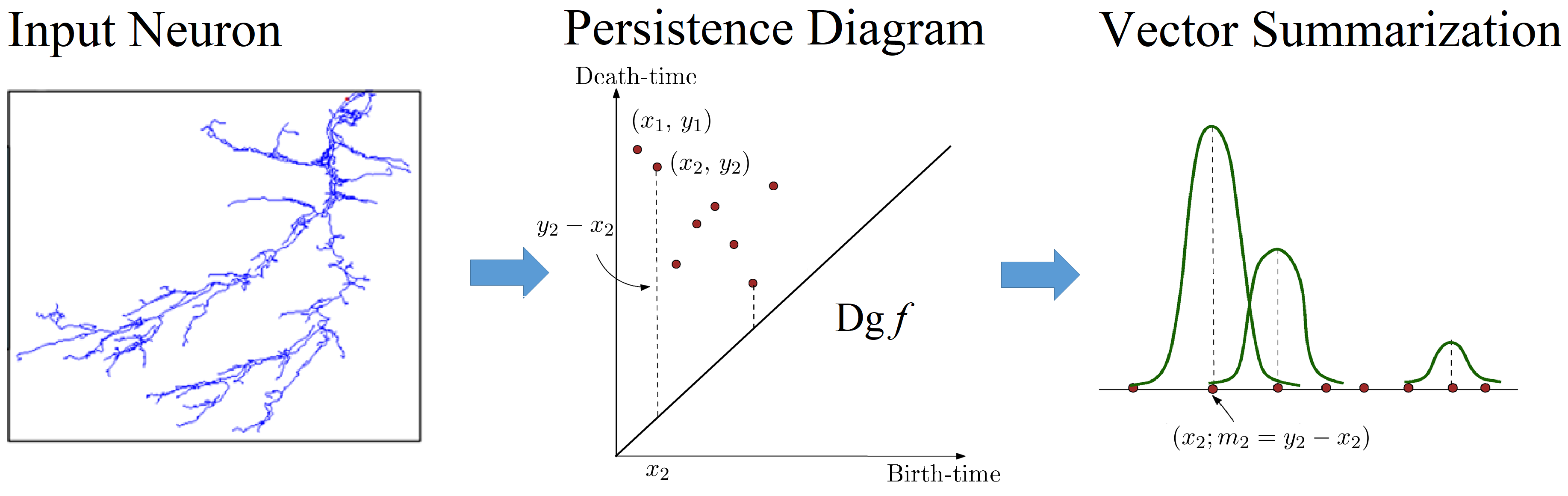
TreePersVec - Feature quantification of morphology data is an important part of determining cell type identity. TreePersVec is a topological based analysis tool used to generate 1D persistence feature vectors of neuron trees. TreePersVec first uses a descriptor function mapping nodes in the neuron tree to positive real values. The default descriptor function uses geodesic distance from any tree node to the root. With function values assigned, TreePersVec will then calculate corresponding persistence diagram and output to a file consisting of a set of 2D points. Coordinates represent the birth and death times of features, and the difference between a feature’s birth and death time shows the importance of the feature. Finally, TreePersVec converts the persistence diagram summary into a 1D persistence feature vector and outputs persistence vector files.. The function for calculating descriptor function values is written in Java, the other functions are written in C++. Source code and instruction are available on GitHub.
Access TreepersVec here:
https://github.com/wangdingkang/NeuronVectorization
Y. Wang Lab (https://web.cse.ohio-state.edu/~wang.1016/), Ohio State University
P. Mitra (https://www.cshl.edu/research/faculty-staff/partha-mitra/), Cold Spring Harbor Lab
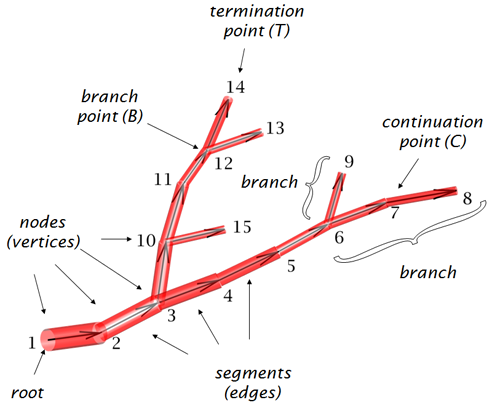
L-Measure is a software tool designed to extract a wide variety of quantitative morphological measurements from neuromorphological reconstructions. Both local parameters (e.g. bifurcation angles) and global descriptors (e.g. total arbor length) can be extracted and combined in many useful analyses, including the popular distributions of surface area as a function of path distance from the soma. Users can specify the target of the analysis by structural domains (e.g. axons vs dendrites) or by morphological features (e.g. terminal branches) . The tool has built-in capability to search for neurons with specific morphological characteristics from a large collection or to compare two neuronal populations with parametric and non-parametric statistical tests. The user-friendly graphical user interface is written in JAVA and can run remotely through a web browser or locally on Linux, Windows, or Mac. The number-crunching engine is written in C++ and can be called from batch scripts for faster execution of large-scale computations. (L-Measure, RRID:SCR_003487)
Access L-Measure here:
http://cng.gmu.edu:8080/Lm/
Related Nature Protocols publication:
https://www.nature.com/articles/nprot.2008.51
G. Ascoli Lab (http://krasnow1.gmu.edu/cn3/ascoli/), George Mason University
BICCN Pipelines
Overview
Pipeline Standards, Maintenance, and Availability
- Open-access and developed with GA4GH standards.
- Written in the Workflow Description Language (WDL), a community-maintained, human-readable workflow language that can run on Cromwell, a portable execution engine that can be launched anywhere, locally or in the cloud.
- Containerized using public Docker instances, allowing researchers to exactly reproduce the workflow software.
- Code is developed and maintained in the WDL Analysis Research Pipelines (WARP) repository in GitHub. Overviews and workflow code for BICCN-related pipelines are linked in the table above; additionally, relevant pipelines can be identified by typing the keyword “BICCN” in the WARP Documentation search bar.The WARP Overview details navigating the repository, pipeline development, and running the workflows.
- Workflows are available for export from Dockstore, a GA4GH-compliant platform for sharing Docker-based tools. Search “warp” on Dockstore to find all WARP pipelines, including those used in the BICCN.
- Workflows are also available to test on Terra, the cloud-based bioinformatics platform used for BCDC data processing. To get started, register for Terra using the registration guide. To try a pipeline, navigate to the pipeline’s workspace linked in the table above or search for the “BICCN” tag in the workspaces tag search bar. Each workspace contains downsampled data, detailed instructions for using the workflows, and cost guidelines. Learn more about Terra with the Getting Started guides.
Citing the Pipelines
Additional Single-cell Transcriptomic Pipeline Resources
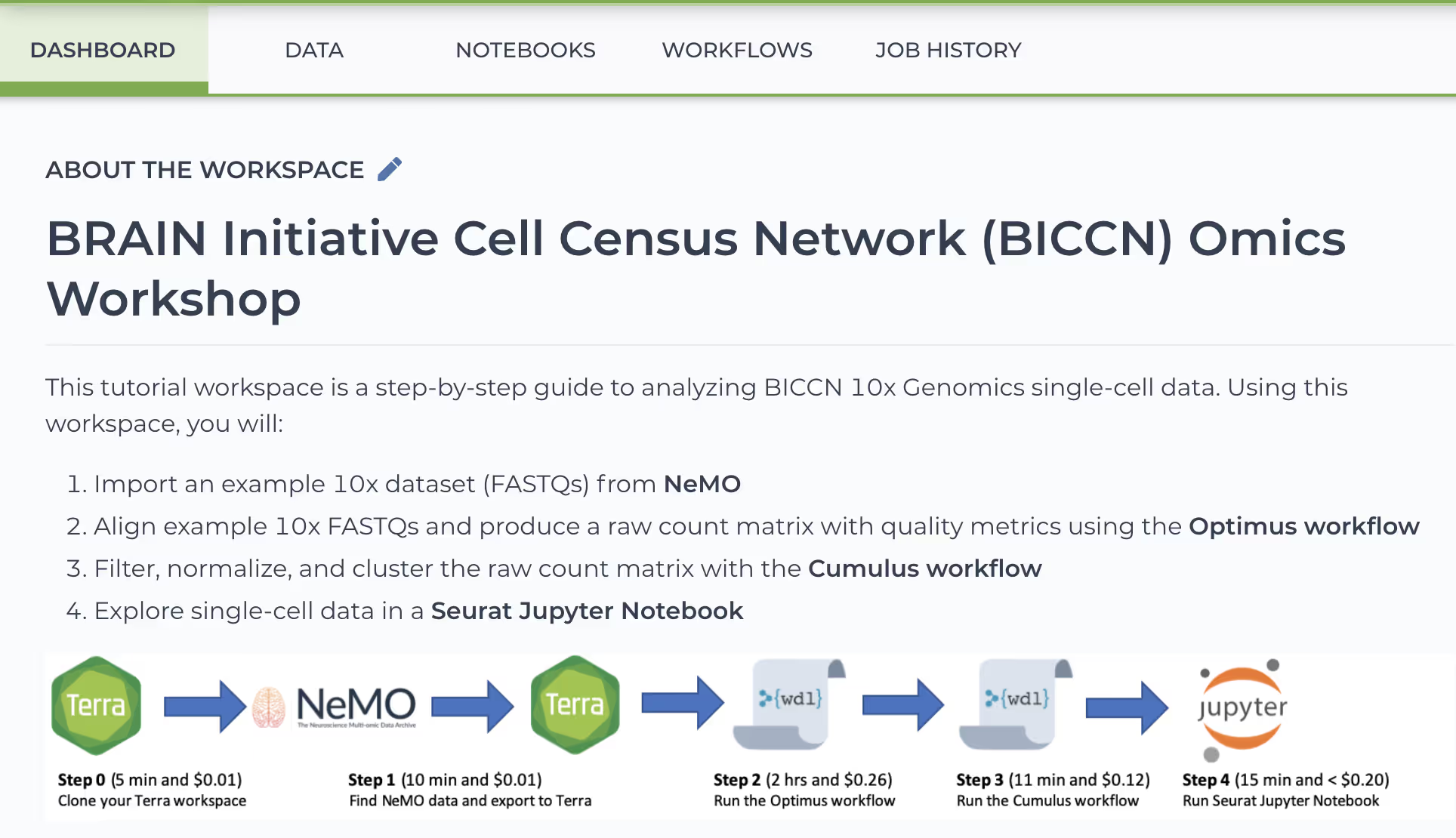
BICCN Omics Workshop Workspace
- Import an example 10x dataset (FASTQs) from NeMO
- Align example 10x FASTQs and produce a raw count matrix with quality metrics using the Optimus workflow
- Filter, normalize, and cluster the raw count matrix with the Cumulus workflow
- Explore single-cell data in a Seurat Jupyter Notebook
BICCN Omics Workshop Webinar Recording
BICCN Omics Workshop Blog
Acknowledgments
