

The NeMO archive and the Terra analysis environment are integrated to assure BICCN data can be analyzed in a secure and scalable environment by any scientist. This integration allows data accessed in NeMO’s multifaceted browser to be quickly handed-off to Terra where data can be analyzed using a secure cloud environment to process and analyze data. This environment spans institutes to promote sharing and has BICCN uniform processing pipelines preinstalled with tutorials. Please read this wiki for a brief overview or watch this BICCN workshop for a live detailed walkthrough.
RRID NeMO Archive: SCR_016152
RRID TERRA: SCR_021648
Generic Web URL NeMO Archive: https://nemoarchive.org
Generic Web URL Terra: https://terra.bio
Contact NeMO Archive: https://nemoarchive.org/contact
Contact Terra: support@terra.bio

Terra is a scalable, open-source platform for biomedical researchers to access data, run analysis tools, and collaborate securely in the cloud. Terra powers important scientific projects including AnVIL, BioData Catalyst, Human Cell Atlas, the BICCN, and many others. Easily access both open and access-controlled datasets hosted in cloud repositories. Explore, analyze, and visualize data using Jupyter Notebooks, RStudio, RShinyApps, and Galaxy. Run your own bioinformatics workflows at scale or try community favorites. Easily share your work with collaborators through secure workspaces.
RRID: SCR_021648
Generic Web URL: https://terra.bio
Contact: support@terra.bio
BICCN Pipelines
Overview
Pipeline Standards, Maintenance, and Availability
- Open-access and developed with GA4GH standards.
- Written in the Workflow Description Language (WDL), a community-maintained, human-readable workflow language that can run on Cromwell, a portable execution engine that can be launched anywhere, locally or in the cloud.
- Containerized using public Docker instances, allowing researchers to exactly reproduce the workflow software.
- Code is developed and maintained in the WDL Analysis Research Pipelines (WARP) repository in GitHub. Overviews and workflow code for BICCN-related pipelines are linked in the table above; additionally, relevant pipelines can be identified by typing the keyword “BICCN” in the WARP Documentation search bar.The WARP Overview details navigating the repository, pipeline development, and running the workflows.
- Workflows are available for export from Dockstore, a GA4GH-compliant platform for sharing Docker-based tools. Search “warp” on Dockstore to find all WARP pipelines, including those used in the BICCN.
- Workflows are also available to test on Terra, the cloud-based bioinformatics platform used for BCDC data processing. To get started, register for Terra using the registration guide. To try a pipeline, navigate to the pipeline’s workspace linked in the table above or search for the “BICCN” tag in the workspaces tag search bar. Each workspace contains downsampled data, detailed instructions for using the workflows, and cost guidelines. Learn more about Terra with the Getting Started guides.
Citing the Pipelines
Additional Single-cell Transcriptomic Pipeline Resources
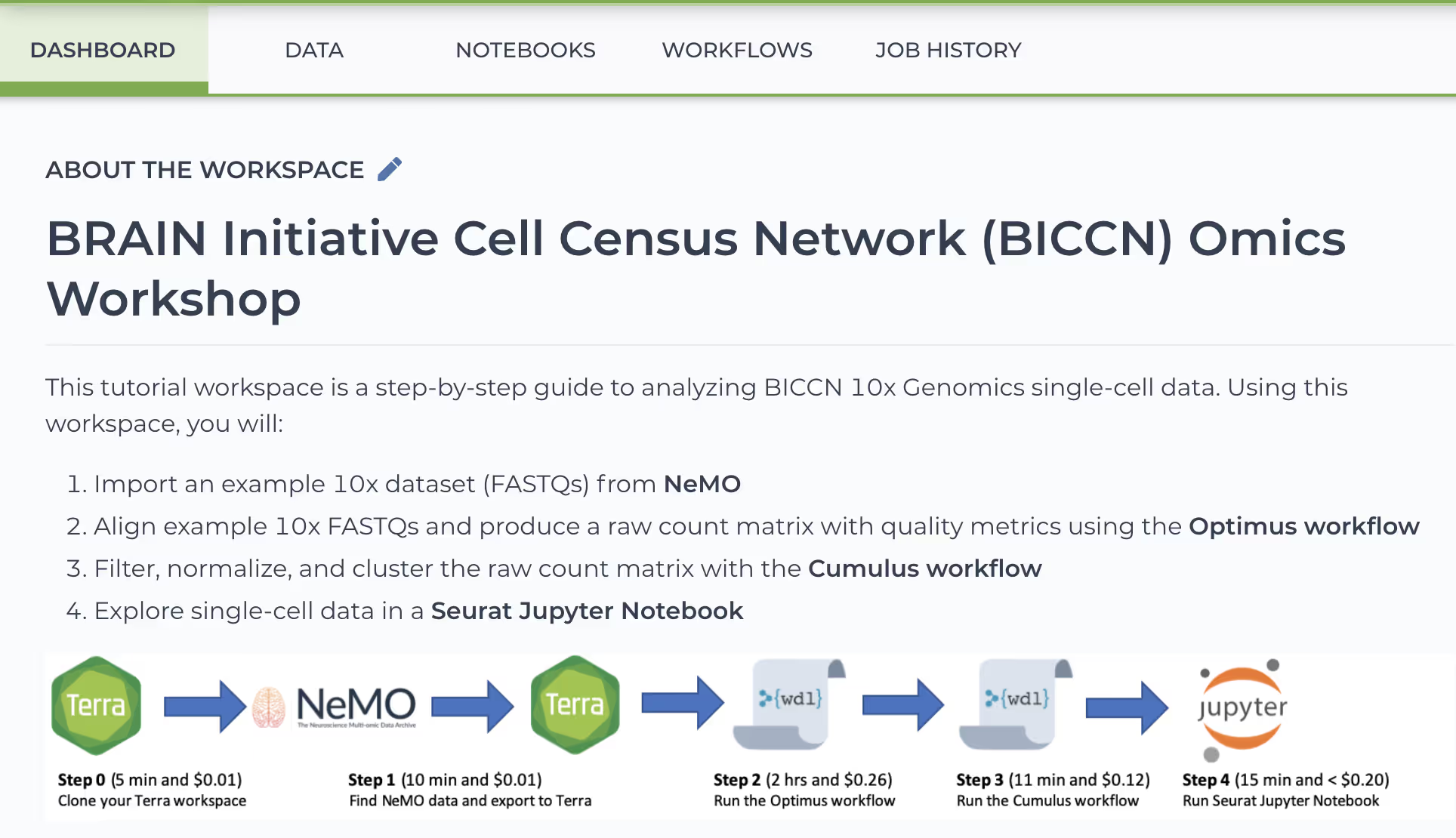
BICCN Omics Workshop Workspace
- Import an example 10x dataset (FASTQs) from NeMO
- Align example 10x FASTQs and produce a raw count matrix with quality metrics using the Optimus workflow
- Filter, normalize, and cluster the raw count matrix with the Cumulus workflow
- Explore single-cell data in a Seurat Jupyter Notebook
BICCN Omics Workshop Webinar Recording
BICCN Omics Workshop Blog
Acknowledgments
